![]()

Chapitre 19
MALADIES AUTO-IMMUNES NON
SPECIFIQUES D’ORGANES :
LES CONNECTIVITES ET LES
VASCULARITES
Les connectivites sont des maladies auto-immunes pouvant toucher tous les tissus de l’organisme. Elles se caractérisent cependant le plus souvent par des lésions cutanées, des douleurs articulaires et une atteinte de l’état général avec syndrome inflammatoire clinique et biologique. Cette triade symptomatique peut être dissociée et un individu peut ne présenter que l’un ou l’autre de ces signes. Ce sont les caractéristiques de la symptomatologie, la présence de certains auto-anticorps et l’éventuelle association avec des atteintes viscérales qui permettent de préciser le diagnostic différentiel entre les connectivites qui seront décrites dans ce chapitre : lupus érythémateux disséminé (LED) ou lupus systémique, dermatomyosite (DM), périartérite noueuse (PAN) ou panartérite noueuse, maladie de Wegener, Sclérodermie, et syndrome de Gougerot-Sjögren.
I – Le lupus érythémateux systémique
Le LES
se caractérise par des atteintes pluri-tissulaires polymorphes, erratiques, mal
systématisées, souvent déconcertantes. Plus les manifestations sont nombreuses,
plus le diagnostic est aisé, mais plus le pronostic est péjoratif, pouvant
aller jusqu'à mettre en jeu la vie du patient. Au contraire, lorsque la maladie
est mono- ou pauci-syptomatique, le diagnostic est hésitant, et il peut errer
plusieurs mois, mais la vie du patient n'est pas menacée en l'absence
d'atteinte d'un organe vital. Des critères ont été définis en 1982, et révisés
en 1997 par l' "American College of Rheumatology", pour le diagnostic
de la maladie (Tableau 1). Il est admis que qu'au moins 4 critères doivent être
présents chez un patient pour que le diagnostic puisse être établi avec certitude.
Dans la majorité des cas le LED survient chez une femme jeune entre 20
et 30 ans. Les trois manifestations cliniques les plus fréquentes sont
articulaires, cutanées et rénales. Chacune de ces atteintes a des
caractéristiques qui permettent de la rattacher à la maladie lupique. La
présence de certains anticorps anti-nucléaires et une diminution du complément
dans le sérum permettent de confirmer le diagnostic.


A – Clinique
A - 1 - L'appareil locomoteur
C'est
l'appareil le plus fréquemment touché, puisqu'une atteinte articulaire ou
osseuse est observée au moins une fois au cours de la maladie chez 95% des
malades. Les atteintes articulaires,
présentes chez 60% des patients, constituent schématiquement 2 tableaux selon
leur allure évolutive:
-
La polyarthrite aiguë se manifeste par l'atteinte fluxionnaire de plusieurs
articulations: surtout les doigts, les poignets, les genoux, les chevilles et
les orteils au début, avec une tendance à la généralisation.
-
La polyarthrite subaiguë, moins inflammatoire, peut devenir chronique et être
confondue avec une polyarthrite rhumatoïde. Les arthrites du LED, contrairement
à celles de la polyarthrite rhumatoïde, n'évoluent pas vers la destruction
articulaire. Elles peuvent en revanche se compliquer de ruptures tendineuses
("rhumatisme de Jaccoud") (Figure 1) entraînant des déformations:
pouce en "z", doigts en "col de cygne", coup de vent
cubital pouvant prêter à confusion avec une polyarthrite rhumatoïde. Les
radiographies permettent de redresser le diagnostic en ne montrant aucune des
destructions propres à cette dernière affection.
Le
LES peut aussi se compliquer d'ostéonécroses
aseptiques indépendamment de la corticothérapie qui peut aussi les
favoriser. Ces infarctus osseux frappent électivement la tête et les condyles
fémoraux, les plateaux tibiaux. Ils peuvent survenir en dehors de toute poussée
de la maladie et se traduisent par une douleur mécanique d'apparition brutale.
La résonance magnétique nucléaire révèle les anomalies plus précocément que la
radiologie standard. Les lésions de vascularite auxquelles on a pu attribuer
l'ostéonécrose du lupus n'ont jamais été observées par les anatomo-pathologistes.
Cette complication, d'autre part, survient souvent en l'absence d'un syndrome
des anti-phospholipides favorisant les thromboses vasculaires: autant dire que
l'on n'en connaît pas l'étiologie.
Les muscles peuvent aussi être affectés
par le LES. Les patients se plaignent de myalgies d'horaire inflammatoire et
l'on constate souvent une élévation modérée des enzymes musculaires dans le
sérum. Lorsqu'elle est pathologique, la biopsie musculaire montre une atrophie
des fibres avec parfois une vascularite et un infiltrat lymphoïde.
Figure 1 : Rhumatisme de
Jaccoud.
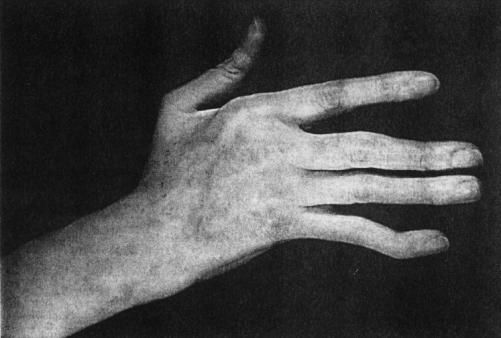
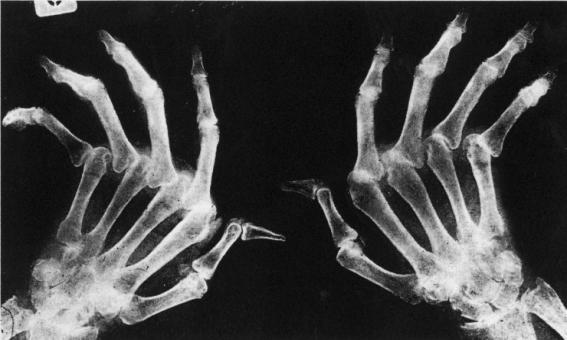
Photo : B Weill
A – 2 - La peau et les muqueuses
Les
signes cutanés varient de l' érythème en ailes de papillon (vespertilio)
siègeant sur les ailes du nez, les pommettes, le front et le menton (15% des
éruptions cutanées) (Figure 2),
aux ulcérations semblables à des morsures de loup (lupus). L'érythème
est déclenché par l'exposition aux rayons ultra-violets B plus qu'aux rayons
ultra-violets A. A cause de cette
photosensibilité, l'éruption ne se limite généralement pas au visage, mais peut
s'étendre à toutes les zones cutanées exposées au soleil.
Environ
15% des malades ayant un LES ne présentent pas de vespertilio, mais un lupus discoïde chronique (LDC) caractérisé par
des lésions papulo-squameuses très infiltrées, à évolution centrifuge, qui
peuvent laisser des cicatrices indélébiles. Il existe aussi des LDC isolés, non
accompagnés de signes systémiques. Seulement 5% d'entre eux évoluent vers le
LES.
Une
forme particulière, le lupus cutané subaigu, s'individualise aussi bien
cliniquement que biologiquement: les lésions érythémato-papuleuses extensives,
souvent squameuses, ont un contour polycyclique et ont tendance à confluer.
Après leur disparition, elles laissent souvent une dépigmentation avec parfois
une atrophie épidermique. Le lupus cutané subaigu est fréquemment associé aux
anticorps anti-Ro/SSA et à un déficit en fraction C2 du complément.
D'autres
lésions non spécifiques du lupus peuvent siéger sur d'autres parties du corps,
notamment les membres. Ce peuvent être des lésions érythémateuses d'apparence
banale ou, plus rarement, des bulles, un érythème polymorphe en cocardes, une
urticaire, des lésions lichénoïdes ou une panniculite.
En
dehors des lésions propres du tissu cutané, la vascularite qui caractérise le
lupus peut toucher les vaisseaux de la peau et entraîner des lésions
nécrotiques. Les lésions, parfois discrètes, punctiformes, traduisent toujours
l'évolutivité de la maladie. Il faut les rechercher au pourtour des ongles et à
la pulpe des doigts et des orteils où elles apparaissent sous forme de taches
purpuriques parfois ulcérées, souvent minuscules.
Des
lésions muqueuses, notamment buccales, mais aussi nasales, génitales et
rectales peuvent être observées. Elles ressemblent à des aphtes mais sont moins
creusantes. Parmi les phanères, ce sont surtout les cheveux qui sont atteints.
Une alopécie en plaque, plus rarement diffuse, peut accompagner les poussées et
régresse après la fin de la poussée.
On
observe le dépôt en bande d'immunoglobulines et de fractions du complément le
long de la membrane basale dermo-épidermique (Figure 3). Ce "Lupus Band
Test" (LBT) est particulièrement caractéristique du LES quand les dépôts
sont constitués d'IgG et de C1q. Le LBT est positif dans plus de 75% des cas
lorsque la biopsie est réalisée en peau pathologique, et dans 50% des cas en
peau saine. La positivité est en faveur de l'évolutivité de la maladie. Des
IgM, du C3 et d'autres fractions du complément peuvent se déposer mais ces
dépôts sont moins caractéristiques du LES.
Figure 2 : Signes cutanés du
LED.
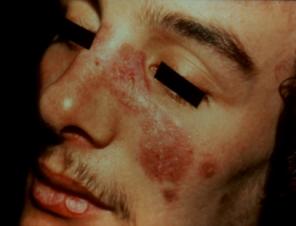
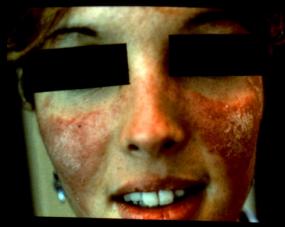
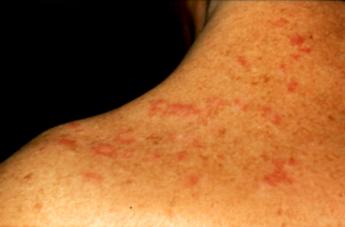
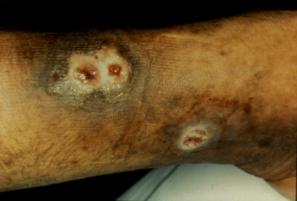
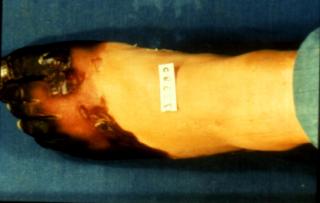
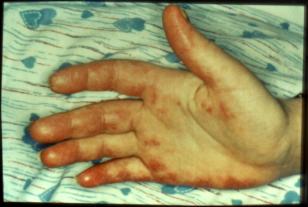
Photo : B Weill
Figure 3 : Aspects de
l’histologie cutanée du LED (Bande lupique).
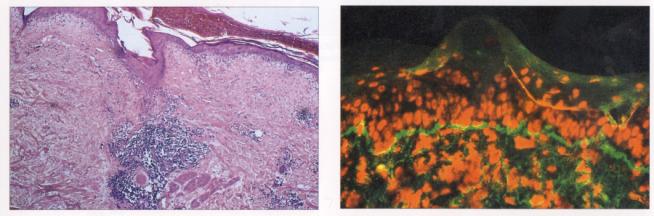
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
A
– 3 - Le rein
L'insuffisance
rénale est rare puisqu'elle ne se manifeste que dans environ 10 % des cas de
LES. En fait, l'atteinte rénale est probablement beaucoup plus fréquente, mais le
plus souvent silencieuse sur le plan clinique, se limitant à une protéinurie
(chez environ la moitié des patients atteints de LES). Lorsqu'elle doit se
manifester, l'atteinte rénale est généralement présente dès la première poussée
et peut révéler la maladie dont elle conditionne le pronostic. Il n'y a pas de
corrélation entre la gravité de l'atteinte générale et celle de la
néphropathie.
L'atteinte
rénale se traduit par une hypertension artérielle, une augmentation de la
créatininémie, une protéinurie et une hématurie microscopique avec une
hyperleucocyturie. Il est utile de la caractériser pour déterminer le
traitement à appliquer.
Une ponction-biopsie rénale est indiquée lorsque le patient a une
hypertension artérielle apparue dans le contexte du lupus ou des signes
biologiques de souffrance rénale : protéinurie ou hématurie non expliquée par
une cause urologique, élévation de la créatinine sérique. Les lésions
histologiques observées après ponction-biopsie rénale sont rarement évolutives.
Elles sont hierarchisées par "classe" et ne changent en principe pas
au cours de l'évolution du lupus, bien que certaines aggravations aient été
observées (Tableau 3)
Figure 4 : Aspect
histologique d’une glomérulonéphrite.
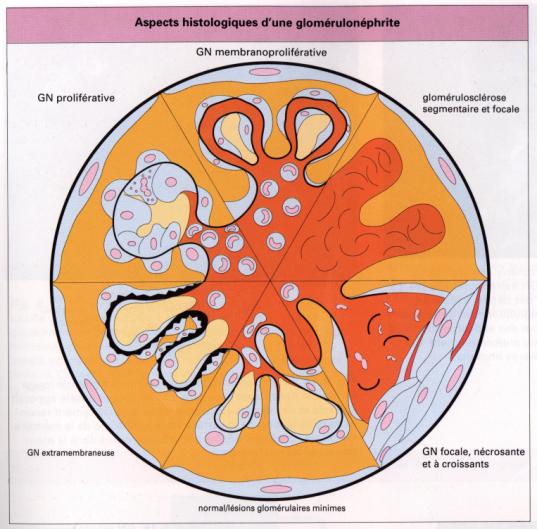
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
Tableau 3 : Atteintes rénales
au cours du LED.
|
Classification morphologique |
Corrélation anatomo-clinique |
|
Classe I :
glomérules normaux A)
Normaux par
toutes les techniques B)
Dépôts en IF
ou en microscopie électronique |
Asymptomatique
ou anomalies minimes (faible protéinurie et hématurie. |
|
Classe
II : Altérations mésangiales A)
épaississement
mésangial ou discrète hypercellularité B)
Hypercellularité
modérée |
A)
Anomalies
urinaires dans 30% des cas B)
Anomalies urinaires
dans 50% des cas |
|
Classe
III : Glomérulonéphrite segmentaire et focale A)
Lésions
nécrosantes actives B)
Lésions
actives et scléreuses C)
Lésions
scléreuses |
Protéinurie
constante, souvent supérieure à 1 g/l Syndrome
néphrotique dans 30% des cas Hématurie Insuffisance
rénale modérée Hypertension
artérielle dans 30% des cas |
|
Classe
IV : Glomérulonéphrite diffuse A)
sans lésions
segmentaires B)
avec lésions
nécrosantes actives C)
avec lésions
actives et sclérosantes D)
avec lésions
sclérosantes |
Protéinurie,
hématurie, leucocyturie constante Syndrome
néphrotique dans 60% des cas Hypertension
artérielle dans 40% des cas Insuffisance
rénale fréquente |
|
Classe V :
Glomérulonéphrite extramembraneuse A)
pure B)
associée à
des lésions de classe II |
Protéinurie très
élevée Syndrome
néphrotique Insuffisance
rénale rare |
|
Classe
VI : Sclérose glomérulaire évoluée |
|
A – 4 - L'appareil respiratoire
L'atteinte la plus fréquente est la pleurésie (50% des cas), uni- ou bilatérale, parfois inaugurale, se
manifestant par de la toux et des douleurs thoraciques. La ponction pleurale
ramène un liquide inflammatoire riche en albumine et en cellules lymphoïdes,
contenant parfois des anticorps anti-nucléaires. Leur détection ne s'impose
généralement pas dans la mesure où ils sont présents avec une plus grande
fréquence dans le sérum.
Plus rarement, on constate sur la radiographie et la
tomodensitométrie du thorax, des infiltrats
interstitiels dont la topographie varie au cours du temps. Les infiltrats
de nature infectieuse sont de loin les plus fréquents au cours du LES. La
nature lupique de lésions pulmonaires (20% des cas) ne doit donc être admise
qu'avec circonspection et lorsque les tests microbiologiques sont négatifs. Les
infiltrats lupiques régressent en général rapidement grâce aux corticoïdes,
mais on peut craindre dans certains cas l'évolution vers une fibrose
interstitielle (5% des cas) et l'insuffisance respiratoire chroniques qui ne
seront plus sensibles à la cortico-thérapie. Les épreuves fonctionnelles
respiratoires montrent un syndrome restrictif et une diminution de la capacité
de transfert de l'oxyde de carbone qui s'aggravent au fil du temps.
Une
hypertension artérielle pulmonaire est de survenue exceptionnelle au cours du
LES.
A – 5 - L'appareil cardio-vasculaire
Le cœur: Des trois tuniques cardiaques,
c'est le péricarde qui est le plus souvent touché (30%) des cas. Comme la
pleurésie, la péricardite peut être latente, n'entraîne pas de tamponnade,
régresse rapidement sous corticoïdes et n'évolue pas vers la constriction. La
myocardite (10% des cas), liée à une vascularite coronaire, est très rare. Elle
se traduit par des troubles du rythme, une tachycardie et finalement par une
insuffisance cardiaque. Depuis l'utilisation des corticoïdes, l'endocardite
verruqueuse de Libman-Sacks est devenue l'exception. Elle touchait
essentiellement les valves mitrales et aortiques.
Les vaisseaux: 20 % environ des
patients éprouvent un syndrome de Raynaud aux doigts, mais aussi aux orteils
et, plus rarement, au nez. Ces troubles vasomoteurs peuvent entraîner une gêne
fonctionnelle, mais habituellement pas de nécrose des extrémités. La
capillaroscopie montre une augmentation de la taille des capillaires mais aucun
signe propre à la sclérodermie.
Les gros troncs vasculaires comme la crosse aortique ne sont en principe
pas atteints. En revanche, surtout si un syndrome des anticorps
anti-phospholipides est associé, des thromboses artérielles ou veineuses,
centrales ou périphériques peuvent survenir.
A – 6 - Le système nerveux
Au
cours du LES, peuvent survenir des atteintes du système nerveux central et du
système nerveux périphérique.
Le système nerveux central: la
manifestation la plus fréquente est la céphalée, parfois d'allure migraineuse
(25% des cas). La deuxième manifestation par ordre de fréquence (20 % des
malades) est la comitialité généralisée, avec des signes
électro-encéphalographiques d'épilepsie essentielle. Cette comitialité est
parfois accompagnée de troubles psychiques. Les crises peuvent être
indépendantes des poussées de lupus.
Des
troubles moteurs d'origine centrale, tels qu'une hémiplégie, une monoplégie, ou
une paraplégie due à une myélite transverse, peuvent apparaître de façon
brutale (15% des cas). Ils accompagnent généralement une poussée de LES et sont
généralement de mauvais pronostic. D'exceptionnelles thrombophlébites du sinus
longitudinal supérieur ont été observées.
Le système nerveux périphérique est
aussi souvent touché (15% des cas). Les atteintes peuvent inclure les nerfs
crâniens comme les nerfs oculo-moteurs. La neuropathie périphérique se traduit
généralement par une multi- ou une mononévrite due à une vascularite des vasa nervorum. Elle peut être confirmée
par une biopsie neuro-musculaire éventuellement guidée par un électromyogramme
préalable.
Les
phénomènes de vascularite responsables de l'ensemble des troubles
neurologiques, aussi bien centraux que périphériques, peuvent faire partie d'un
syndrome des anticorps anti-phospholipides; dans ce cas, on pourra trouver des
anticorps anti-cardiolipine et anti-bêta 2-GP1 dans le sérum.
Un syndrome méningé peut survenir
exceptionnellement à l'occasion d'une poussée. Le LCR est inflammatoire, avec essentiellement
des éléments lymphocytaires. Le diagnostic différentiel avec une origine
infectieuse peut être difficile. La normalité du taux de CRP sérique est en
faveur d'une méningite purement inflammatoire entrant dans le cadre de la
maladie lupique.
Le LES peut se compliquer de manifestations psychiatriques isolées
qui inaugurent parfois la maladie. Il peut s'agir d'un syndrome dépressif, d'un
délire, d'une désorientation, d'hallucinations ou d'une psychose paranoïde ou
schizoïde. En l'absence d'autres signes cliniques évocateurs de la maladie, ou
de syndrome inflammatoire, le diagnostic peut être très difficile.
Il
faut inclure dans les atteintes neurologiques, les complications oculaires. Ce sont, avant tout, des
conjonctivites et des épisclérites, mais, outre des paralysies oculo-motrices
dues à une neuropathie périphérique, on peut observer une vascularite
rétinienne. Elle se traduit par des exsudats cotonneux typiques de la rétinite
dysorique. Ils sont non spécifiques du LES et sont souvent associés à des
hémorragies (5% des cas). Plus rarement, le fond d'œil révèle une thrombose de
l'artère centrale de la rétine ou d'une artère cilio-rétinienne.
Figure 5 : Atteinte
occulaire du LED.
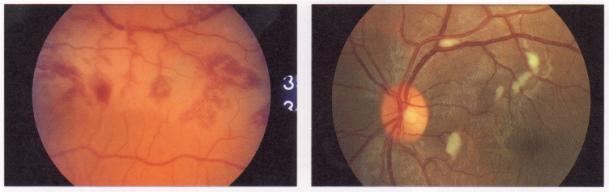
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
A
- 7 - le foie et l'appareil digestif:
Le foie: Le diagnostic différentiel
entre une hépatite auto-immune et une hépatite lupique est parfois très
difficile. Les signes cliniques et biologiques de la cytolyse hépatique sont
les mêmes quelle que soit l'étiologie. Une élévation isolée des transaminases
sériques est observée chez 40% des malades. L'association d'autres signes
cliniques caractéristiques du LES, comme une atteinte cutanée ou rénale, peut
permettre de clarifier le diagnostic. En leur absence, les investigations
biologiques peuvent aussi orienter vers une étiologie: une hépatite auto-immune
de type I s'accompagne d'anticorps anti-muscle lisse, une hépatite auto-immune
de type II, d'anticorps anti-LKM-1. Dans les deux cas on peut détecter aussi
des anticorps anti-nucléaires, mais en principe jamais d'anticorps anti-ADNn ni
d'anticorps anti-Sm. Au contraire, au cours des atteintes hépatiques du LES ces
derniers auto-anticorps sont souvent détectés, et si les anticorps caractéristiques des hépatites
auto-immunes sont présents, c'est en général à des taux très modérés.
L'histologie hépatique n'est pas toujours d'une grande aide pour le diagnostic
différentiel.
Le tube digestif: les atteintes du tube
digestif au cours du LES sont plus souvent d'origine iatrogénique, liées à la
prise d'anti-inflammatoires, que spécifiques de la maladie. Les malades
éprouvent dans 30% des cas des nausées, des douleurs abdominales et de la
diarrhée. Les symptomes liés à une vascularite sont très rares et variables
selon la topographie. En cas d'atteinte d'un gros tronc artériel, un syndrome
pseudo-chirurgical peut survenir, aboutissant dans des cas exceptionnels, à une
perforation intestinale. Une microvascularite peut entraîner un syndrome de
malabsorption.
A – 7 - les organes lymphoïdes
Une
splénomégalie et des adénomégalies sont palpées dans 15% des cas.
B - Biologie
On
peut distinguer les examens biologiques permettant de poser le diagnostic de LES
et ceux qui permettent d'apprécier l'évolutivité de la maladie (Figure 4 et
Tableau 4). Les examens complémentaires permettant de déceler les complications
viscérales sont propres à chaque atteinte et ne sont pas envisagés ici.
Le premier examen biologique qui a permis de caractériser la maladie
lupique est la formation in vitro de cellules LE. Bien que cet examen soit
aujourd'hui obsolète et remplacé par la détection des anticorps anti-nucléaires
(AAN), les cellules LE reflètent un phénomène physiopathologique probablement à
l'origine des poussées de LES. Le phénomène LE représente en effet la
phagocyose de cellules en apoptose, qui déclenche la réaction auto-immunitaire
aboutissant à la production d'auto-anticorps anti-nucléaires et aux
manifestations cliniques de la maladie .
B – 1 - Les auto-anticorps anti-nucléaires (AAN)
Les
AAN sont les marqueurs sériques les plus caractéristiques du LES. Ce sont des
immunoglobulines spécifiques de différents composants nucléaires : acides
nucléiques, histones, ribonucléoprotéines. Leur détection globale est réalisée
en général par un test d'immunofluorescence indirecte sur un frottis cellules
HEp-2, cellules malignes humaines possédant un noyau volumineux et donc
particulièrement propices à la détection des AAN. Les résultats sont rendus en
titres d'anticorps (Figure 5).
L'interprétation
d'une recherche d'AAN peut être difficile: d'une part, la présence d'AAN ne
traduit pas toujours une maladie auto-immune. L'auto-immunité ne procède pas du
mode "tout ou rien", et certains AAN (surtout lorsqu'ils sont de
l'isotype IgM) peuvent être détectés chez des sujets ayant un syndrome
inflammatoire non auto-immun. D'autre part, l'interprétation peut être rendue
difficile par la faiblesse d'un titre d'anticorps. Un taux bas d'AAN peut
n'avoir aucune signification pathologique chez un adulte, surtout s'il a plus
de 70 ans. Un faible taux d' AAN
peut cependant traduire dans certains cas une maladie débutante et, chez
l'enfant, il est rarement dépourvu de signification pathologique. La présence
d'AAN dans un sérum doit donc être interprétée en fonction de la clinique car,
si des AAN sont détectés chez 99% des patients ayant une poussée de LES, de
nombreuses autres maladies inflammatoires auto-immunes ou non peuvent aussi en
comporter.
Pour
permettre une interprétation du résultat des AAN, la confrontation
clinico-biologique doit être doublée d'une détermination des spécificités de
ces anticorps. Seules certaines spécificités permettent d'affirmer presque à tout
coup le diagnostic de LES : ce sont les anticorps anti-ADN natif (AANn) ou à
double brin, et les anticorps anti-Sm.
L' aspect de la fluorescence nucléaire des cellules HEp-2 peut être
évocatrice: les anticorps anti-désoxyribonucléoprotéines (notamment
anti-histones) confèrent une fluorescence homogène, les anticorps anti-ADNn une
fluorescence périphérique et les anticorps anti-ribonucléoprotéines (dont les
anticorps anti-Sm) une fluorescence mouchetée. D'autres aspects ont été décrits
mais aucun aspect, même typique,
ne dispense de la réalisation de tests complémentaires qui, seuls,
permettent de caractériser avec certitude la spécificité des anticorps (Tableau
5).
B
– 1 – 1 - Les anticorps anti-ADN
natif :
Leur dosage est indispensable en cas de positivité des AAN car leur
présence témoigne généralement que le patient est atteint d'un LES. En effet,
70% des malades ont au moins une fois des anticorps anti-ADNn au cours de
l'évolution de leur maladie. La présence d'anticorps anti-ADNn est en effet exceptionnelle
au cours d'autres affections que le LES.
Etant
donné le caractère capital de leur découverte pour le diagnostic et le
pronostic, les anticorps anti-ADNn doivent être détectés par deux techniques
reposant sur des principes méthodologiques différents. Les trois méthodes les
plus couramment utilisées sont l'immunofluorescence indirecte sur Crithidia
luciliae, la radio-immunologie (test de Farr) et les dosages
immuno-enzymatiques (ELISA).
Seuls
les anticorps anti-ADNn de forte affinité, c'est-à-dire généralement les IgG,
sont caractéristiques du LES. Ces seuls anticorps sont détectés sur Crithidia
luciliae et par le test de Farr. Les tests ELISA, en revanche, détectent aussi
les anticorps IgM de faible affinité non caractéristiques du LES. Il est donc
indispensable, lorsque les anticorps anti-ADNn sont recherchés par ELISA, de
doser séparément les IgM et les IgG qui, seules, permettent de poser le
diagnostic de LES.
On
peut en effet rencontrer des IgM anti ADNn au cours d'autres connectivites comme
la polyarthrite rhumatoïde ou au cours d'infections virales comme les
hépatites. Une forte concentration d'IgM anti-ADNn peut même entraîner,
parfois, une faible positivité du test sur Crithidia luciliae.
Contrairement
à la détection globale des AAN, la concentration des anticorps anti ADNn peut
apporter des renseignements sur l'évolutivité du LES. Une augmentation rapide
du titre des anticorps anti-ADNn traduit généralement l'évolutivité de la
maladie et doit faire craindre une atteinte viscérale. Un taux élevé mais
stable n'a pas cette valeur indicative. Il faut noter que pour apprécier
l'évolutivité, la clinique et le taux du complément sont plus fiables que le
titre des anticorps anti ADNn.
B
– 1 – 2 - Les anticorps
spécifiques d'antigènes nucléaires solubles
Ces
anticorps reconnaissent des épitopes peptidiques constitutifs de molécules
ribonucléoprotéiques. Ils sont couramment recherchés par immunoprécipitation en
gélose selon la technique d'Ouchterlony ou par contre-immuno-électrophorèse
(électrosynérèse) en utilisant un extrait de cellules thymiques de lapin (ECT)
comme substrat. La détection par immuno-empreinte ("Western blot")
procure des résultats parfois difficiles à interpréter et n'est donc pas
utilisée pour le diagnostic médical. Des techniques immuno-enzymatiques sont en cours de développement.
Les auto-antigènes ribonucléoprotéiques reconnus par les anticorps
anti-ECT sont constitués de chaînes d'ARN U1, U2, U4, U5 ou U6 liées à des
molécules protéiques A, B/B', C, D, E, F, G et à une molécule de 68 kDa.
Les anticorps anti-Sm se lient aux
protéines B/B' D, E, F, G communes aux 5 chaînes d'ARN. Les anticorps anti-Sm,
exceptionnellement trouvés en dehors du LES, sont aussi caractéristiques de
cette maladie que les anticorps anti-ADNn. En revanche, ils sont beaucoup moins
souvent positifs (Tableau 4). En outre, contrairement aux anticorps anti-ADNn,
leur concentration ne reflète ni un risque d'atteinte viscérale, ni
l'évolutivité du LES.
Les anticorps
anti-U1 RNP reconnaissent la protéine de 68 kDa et les protéines A et
C liées à la chaîne ARN U1. Ces anticorps, initialement décrits dans la
connectivite mixte, sont fréquents dans le LES mais n'en sont pas spécifiques.
On peut aussi les détecter au cours de la polyarthrite rhumatoïde, la
polymyosite, la sclérodermie systémique, et au cours des lupus médicamenteux.
Les
anticorps anti-Ro/SSA : Parmi les anticorps anti-ribo-nucléoprotéines figurent
aussi les anticorps anti Ro/SSA. Comme les antigènes Ro/SSA sont peu
représentés dans le thymus de lapin, on utilise généralement la rate humaine
comme substrat pour leur détection. Les anticorps anti Ro/SSA reconnaissent
soit une protéine de 52 kDa, soit une protéine de 60 kDa fixée sur une chaîne
d'ARN, sans que des différences dans la présentation clinique ou les
complications soient associées à l'une de ces deux spécificités. Ils sont
présents dans 30% des LES, mais peuvent être observés dans la polyarthrite
rhumatoïde et surtout le syndrome de Gougerot-Sjögren où ils ont été décrits.
Chez
1% des patientes atteints de LES,
on ne décèle pas d'AAN par immunofluorescence indirecte. Il semble que
le pourcentage ait diminué depuis l'utilisation des cellules HEp-2, mais de
telles situations s'observent toujours. Dans ces cas, on trouve généralement
des anticorps anti-Ro/SSA par immunoprécipitation. Ils correspondent à des formes subaigues de LES comportant une
atteinte cutanée extensive, parfois généralisée, avec une très grande
photosensibilité. La présence d'anticorps anti-Ro/SSA est fréquemment associée
à un déficit congénital en fraction C2 ou C4 du complément.
En
outre, il est indispensable de rechercher les anticorps anti-Ro/SSA chez toute femme enceinte atteinte d'une
connectivite car ces anticorps peuvent, dans 5% des cas, être pathogènes pour
le myocarde foetal et entraîner un bloc auriculo-ventriculaire congénital. En
effet, les myocytes foetaux expriment à leur surface des molécules de Ro (52 et
60 kDa) sur lesquelles peuvent se fixer des IgG maternelles anti-Ro qui ont
franchi la barrière placentaire. Cette fixation peut entraîner un
bloc-auriculo-ventriculaire congénital. La présence d'anticorps anti-Ro/SSA
chez une femme enceinte rend donc nécessaire une surveillance cardiologique du
foetus et un accouchement dans un milieu obstétrical apte à donner les soins
requis à la naissance.
Les anticorps anti-La/SSB reconnaissent
une protéine de 47 kD fixée sur une chaîne d'ARN. Ils ne sont présents que dans
10% des LES et sont toujours associés à un anticorps anti-Ro/SSA, sans que la
réciproque soit vraie.
Autres anticorps anti-nucléaires : des
anticorps anti-PCNA ("Proliferating Cell Nuclear Antigen") sont
détectés chez moins de 10% des malades atteints de LES. Ils reconnaissent une
protéine auxiliaire d'ADN polymérase et caractérisent des formes graves de la
maladie, avec atteintes rénale et neurologique fréquentes. De nombreux autres
AAN peuvent être détectés aucours du LES, mais leur valeur diagnostique est
faible.
B – 2 - Les anticorps
anti-phospholipides
Le
syndrome des anticorps anti-phospholipides a été initialement décrit par
Soulier et Boffa en 1981. Il se caractérise par des avortements à répétition, des thromboses
veineuses et artérielles centrales et périphériques.
L'association
possible de ce syndrome à un LES, explique en partie les complications
obstétricales observées au cours du lupus ( Tableau 6).
Cette
association explique ce que l'on appelait autrefois la "fausse
positivité" de la sérologie syphilitique. Au cours du syndrome des
anticorps anti-phospholipides apparaissent en effet des anticorps anti-cardiolipine responsables de la
positivité des réactions sérologiques de la syphilis utilisant la cardiolipine
comme substrat. Ces anticorps peuvent, en outre, reconnaître d'autres
phospholipides comme la la phosphatidylsérine (proche de la cardiolipine), la
phosphatidyléthanolamine, l'acide phosphatidique et le phosphatidylanositol. Ce
sont cependant les anticorps anti-cardiolipine qui sont les plus constamment
présents au cours du syndrome des anti-phospholipides, et c'est eux qu'il
convient de rechercher à des fins diagnostiques.
Les
anticorps anti-phospholipides peuvent, en outre, se fixer sur certaines enzymes
de la coagulation comme le facteur VIII, et exercer in vitro une activité
anti-coagulante. Cette activité se traduit par un allongement du temps de
céphaline-kaolin que l'on ne corrige pas par l'addition de plasma normal.
L'expression "anticoagulant du lupus" est erronée puisqu'in vivo, ces
anticorps entraînent au contraire des thromboses.
Les anticorps anti-phospholipides peuvent aussi se fixer sur les plaquettes et
entraîner une thrombopénie. Ils sont
dosés par ELISA en présence d'un cofacteur, la bêta 2-GP1 apportée par le sérum
utilisé pour saturer les puits de la plaque de microtitration. En présence de
la bêta 2-GP1, la cardiolipine
forme un complexe reconnu par des anticorps polyclonaux de spécificité
variable. Certains ne reconnaissent que la cardiolipine. Ils ne sont pas
spécifiques du syndrome des anticorps anti-phospholipides et peuvent apparaître
au cours de syndromes inflammatoires variés comme les infections virales, la
cirrhose, la sarcoïdose et certains cancers. En revanche, les anticorps qui
reconnaissent un épitope conformationnel du complexe cardiolipine - bêta 2-GP1
sont très spécifiques du syndrome des anticorps anti-phospholipides associé ou
non à un LES.
B – 3 – Le complément
sérique
Au
cours du LES, le CH50 peut être
abaissé de façon permanente à cause d'un déficit congénital en C2 ou en C4, ou
de façon transitoire à cause d'une consommation de certaines fractions du
complément lors des poussées de la maladie.
Un
patient sur deux a un déficit
hétérozygote en C2 ou en C4. Dans ces cas, le CH50 est constamment abaissé,
généralement autour de la limite inférieure de la normale. Ces déficits sont
associés aux gènes HLA A1, B8 et DR3. En l'absence d'un déficit congénital, le
CH50 est normal en dehors des poussées.
En
revanche, le LES est la seule connectivite où le CH50 est abaissé pendant les poussées. L'abaissement du CH50 est
dû à une consommation de C3, de C4, et souvent de facteur B, traduisant
l'activation du complément par les deux voies directe et alterne.
Lorsque
le LES est évolutif, on observe donc simultanément une diminution du taux
sérique de CH50, C3, de C4 et de facteur B.
B – 4 – La vitesse de
sédimentation globulaire
Elle
est accélérée pendant les poussées et revient, en principe, à la normale pendant les phases de
rémission. Une accélération de la VS n'est nullement caractéristique du LES,
mais peut traduire un syndrome inflammatoire de n'importe quelle origine.
La
concentration plasmatique de la "C Réactive Protein" (CRP), n'est
jamais augmentée au cours du LES, même pendant les poussées. Une augmentation
de la CRP au cours d'un LES doit faire suspecter une complication infectieuse.
B – 5 – La
numération-Formule sanguine
On
constate très souvent une anémie au
cours de la maladie. Elle peut avoir plusieurs causes :
-
normochrome, normocytaire et hyposidérémique, l'anémie reflète le syndrome inflammatoire.
-
hypochrome, microcytaire et hyposidérémique, l'anémie peut traduire un saignement d'origine digestive
consécutif aux traitements anti-inflammatoires. Elle est généralement modérée.
-
associée à une réticulocytose, elle suggère une hémolyse et doit faire prescrire un test de Coombs. En fait, les anémies hémolytiques sont rares au
cours du LES, ou alors elles se manifestent d'emblée, dans le cadre d'un
syndrome d'Evans. En revanche le test de Coombs est positif dans 20 à 25 % des
cas, même en l'absence d'hémolyse, révèlant la fixation d'IgG autologues et de
complément sur les hématies.
Une
leucopénie, portant sur les lignées
granuleuse et lymphocytaire, est très fréquemment observée. Son origine n'est
pas univoque et peut être centrale ou périphérique, liée dans ce dernier cas à
la production d'auto-anticorps anti-polynucléaires et anti-lymphocytes.
De
même, des anticorps anti-plaquettes peuvent expliquer la survenue d'un purpura thrombopénique parfois
inaugural.
B – 6 – Electrophorèse
des protides
Il
montre, surtout pendant les poussées, une hypergammaglobulinémie
polyclonale qui n'a rien de spécifique du LES.
Figure 6 : Biologie du Lupus
érythémateux systémique.
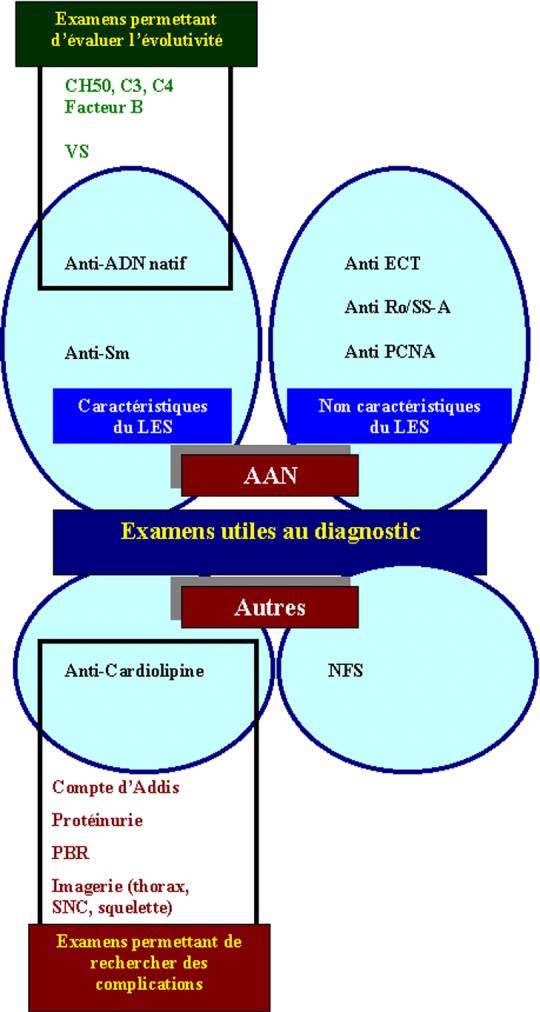
Figure 7 : Algorythme
diagnostic.
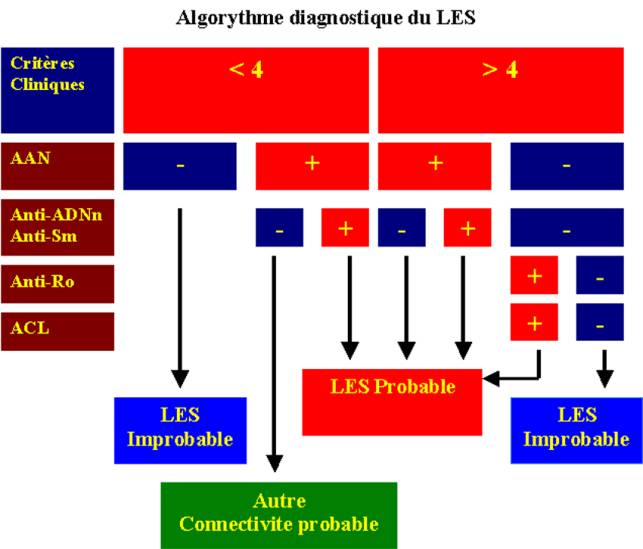
C - Physiopathologie
Bien que
plus de mille publications soient consacrées chaque année à la recherche sur la
physiopathologie du LES, on compte encore dans ce domaine plus d'incertitudes que
de connaissances assurées. Il est cependant établi aujourd'hui que la maladie
résulte de la rupture de la tolérance naturelle vis-à-vis d'une série d'
épitopes présents au sein des nucléosomes. Cette auto-immunisation est
favorisée à la fois par le terrain génétique et par la survenue d' une
agression capable d'induire la mort des cellules cibles par apoptose, comme des radiations ultra-violettes ou
un agent infectieux.
Au
cours de la mort cellulaire par apoptose, un des premiers phénomènes observés est
la fragmentation de la chromatine qui entraîne la production de nucléosomes
constitués de molécules d'histones (H1, H2A, H2B, H3 et H4) entourées d'un
double brin de 150 à 180 paires de bases d'ADN enroulé selon deux tours de spire. Très rapidement, les
nucléosomes sont exprimés à la surface de la cellule en apoptose. Certains vont
être relargués dans le milieu extérieur, d'autres vont être phagocytés avec la
cellule apoptotique par des macrophages.
Les
macrophages vont traiter les différents auto-antigènes de la cellule phagocytée
et présenter à leur surface, par l'intermédiaire de leurs molécules HLA de
classe II, des peptides dérivés
des histones nucléosomiales. Ces peptides sont reconnus par des lymphocytes T
CD4+ auto-réactifs. La phagocytose des cellules en apoptose est en
outre suivie d'une forte production d'IL-6 et d'IL-10 par les macrophages. Dans
le contexte inflammatoire lié à l'infection ou à l'agression physique, la
reconnaissance des auto-antigènes est capable de lever leur anergie et l'environnement
cytokinique favorise leur différenciation en lymphocytes TH-2.
D'autre
part, les nucléosomes libérés dans le milieu extérieur peuvent être captés par
le récepteur d'antigène de certains lymphocytes B (BcR) auto-réactifs, comme
par exemple des lymphocytes B reconnaissant l'ADN natif constitutif des
nucléosomes. Les lymphocytes B se comportent comme des CPA, endocytent les
nucléosomes, les traitent, et exposent des peptides d'histones sur leurs
molécules HLA de classe II membranaires. Des lymphocytes TH-2 spécifiques des
peptides d'histone les reconnaissent et induisent la différenciation des
lymphocytes B en plaSmocytes producteurs d'anticorps. Les anticorps produits
sont des auto-anticorps spécifiques de l'antigène reconnu par le BcR, c'est-à-dire
de l'ADN natif. C'est ainsi que des nucléosomes produits par des cellules en
apoptose engendrent d'une part une réaction auto-immune de type TH-2 dirigée
contre des histones (essentiellement H2A et H2B), et d'autre part la production
d'anticorps anti-ADN natif par des lymphocytes B stimulés par les lymphocytes
TH-2 anti-histones (Figure 6). Les mécaniSmes de production des autres
auto-anticorps est probablement assez semblable car les autres auto-antigènes
nucléaires sont aussi exposés à la surface des cellules apoptotiques.
Ni
les lymphocytes TH-2 ni les anticorps anti-ADN ne sont directement pathogènes.
Ce sont les complexes formés entre les auto-anticorps et les auto-antigènes
libérés lors de l'apoptose, qui
induisent des phénomènes inflammatoires par l'intermédiaire de l'activation du
complément dans les tissus où ils se déposent. Ceci explique la chute du CH50
et la consommation des fractions du complément observées au cours des poussées
du LES. Le fréquent déficit hétérozygote en C4 diminue les capacités des
malades d'éliminer les complexes immuns et augmente le risque d'inflammation
tissulaire.
Lors
d'une poussée ultérieure, dont l'élément déclenchant reste à préciser, les
auto-anticorps seront prêts à se combiner avec les auto-antigènes fraîchement
libérés et à former des complexes délétères pour les tissus. D'autre part, les
lymphocytes TH-2 auto-réactifs à mémoire stimuleront de manière accélérée de
nouveaux lymphocytes B pour leur faire produire de nouveaux anticorps dont
l'ascension pourra être constatée dans le sérum.
Si
les phénomènes d'apoptose cellulaire n'entraînent pas la survenue d'un lupus
systémique chez tous les individus, c'est que le patrimoine génétique joue un
rôle dans le déclenchement des phénomènes d'auto-immunisation. La présentation
des auto-antigènes par les molécules HLA des CPA est déterminée qualitativement
et quantitativement par l'affinité de ces molécules entre elles. Certaines
molécules HLA sont beaucoup plus efficaces que d'autres pour présenter des
auto-antigènes, et les sujets qui en sont porteurs sont prédisposés aux
maladies auto-immunes.
Le
lupus systémique peut donc être considéré comme une vascularite systémique
provoquée par des complexes immuns. Les constituants auto-antigéniques de ces
complexes résultent d'une réaction auto-immunitaire de type TH-2 vis-à-vis de
composants nucléosomiaux rendus
accessibles lors d'une apoptose cellulaire massive.
D - Evolution
- Spontanément, la maladie est rarement aiguë, fébrile, mortelle en quelques mois.
- Plus souvent, elle se fait par poussées déclenchées ou exacerbées par l'insolation, la grossesse, la diminution intempestive des corticoides; les poussées alternent avec des rémissions plus ou moins complètes, obtenues par le traitement, mais parfois spontanées.
- Le traitement a transformé le pronostic, et, en l'absence d'atteinte rénale, la survie peut dépasser 20 ans ou plus.
E - Il faut
signaler certains problèmes particuliers posés par
1/ Le lupus érythémateux chronique, uniquement cutané, ne met pas en jeu le pronostic vital; le L.E. subaigu associe des signes cutanés voisins du lupus discoïde et des signes viscéraux et biologiques rappelant ceux du L.E.D. mais de gravité moindre.
2/ Le diagnostic différentiel avec la dermatomyosite peut être très difficile; l’association possible des deux maladies peut encore compliquer la question
3/ La polyarthrite rhumatoïde pose aussi des problèmes nosologiques avant l’apparition des lésions articulaires caractéristiques. Elle peut aussi être aasociée au LED.
4/ Les lupus induits par les médicaments (Au, Bi, As, Péni., Sulfamides, INH, Hydantoïnes, Hydralazines, Procaïnamide, d-pénicillamine) peuvent simuler cliniquement et biologiquement un LED. Ils ne comportent cependant jamais ni Ac anti-ADN natif ni Ac anti-Sm. (cf Chapitre sur les "auto-anticorps non spécifiques d’organes").
5/ L'étiologie virale du lupus fut un temps envisagée, en raison de la présence d'inclusions virales dans la peau ou le rein des malades. En fait, aucune preuve irréfutable n'a pu encore être apportée.
F -
Traitement
1/ Il est basé sur la corticothérapie, dont la posologie dépend du degré d'extension viscérale, et en particulier du type histopathologique de l'atteinte rénale. Les doses d'attaque doivent être fortes, et diminuées très progressivement jusqu'à une dose d'entretien longtemps poursuivie. On peut ,au début, utiliser les bolus i.v. de Solumédrol dans les formes sévères.
2/ Les anti-paludiques ont un rôle plus modeste dans ce lupus systémique; insuffisants à arrèter une poussée, ils sont cependant associés à titre de traitement de longue durée et peuvent suffire dans les formes bénignes.
3/ Les immunodépresseurs, anti-métabolites (Imurel), moutardes à l'azote (Endoxan) pourront être discutés en cas de néphropathie ou de vascularite sévère, en surveillant la formule sanguine.
II – Les myopathies inflammatoires ou
myosites
Les
myopathies inflammatoires comprennent la polymyosite (PM), la dermatomyosite
(DM) dont l'incidence globale dans la population européenne occidentale est de 10 / 106 et la
prévalence de 5 / 105. Les myosites sporadiques à inclusions (MI)
sont beaucoup plus rares. Les myosites sont les connectivites les plus
fréquentes chez l'enfant. Bien que les dermatomyosites se manifestent sur un
mode souvent aigu et les polymyosites sur un mode chronique, toutes les
myosites ont en commun des atteintes musculaire, articulaire et respiratoire.
Ces atteintes seront d'abord étudiées dans le cadre des polymyosites où elles
restent isolées. Les atteintes cutanées de la dermatomyosite seront ensuite
décrites, ainsi que les particularités de l'atteinte musculaire au cours des
myosites à inclusions.
A
- Signes cliniques des polymyosites
Les myosites
correspondent à une inflammation de cause inconnue des muscles striés. Cette
atteinte se caractérise par des myalgies accompagnées d’une faiblesse
musculaire symétrique qui prédomine aux ceintures scapulaire et pelvienne, au
pharynx et à la sangle abdominale et s’étend progressivement en respectant les
muscles lisses. La pression des muscles est douloureuse, leur consistance prend
celle du carton et entraîne une limitation des mouvements.
Le myocarde peut être atteint, de manière le plus
souvent asymptomatique: dans plus de 30% des cas on note des anomalies
électrocardiographiques telles que des troubles de la conduction (blocs de
branche, blocs auriculo-ventriculares, troubles de la repolarisation), mais les
manifestations cliniques correspondant à ces troubles électriques sont très
rares. La myocardite peut cependant évoluer vers la fibrose et l'insuffisance
cardiaque.
Dans 20% des cas les malades éprouvent des
arthralgies inflammatoires touchant préférentiellement les poignets, les
genoux, les épaules et les mains. On connaît des formes enraidissantes avec ou
sans syndrome inflammatoire.
Une atteinte respiratoire peur survenir au cours des
myosites. Le déficit musculaire peut en effet entraîner une hypoventilation
responsable de dyspnée. Cependant, dans 10% des cas, une toux et une dyspnée
accompagnées de fièvres et d'images réticulo-nodulaires à la radiographie,
résultent d'une pneumopathie interstitielle diffuse. Dans le cadre du
"syndrome des anti-synthétases", la myosite et la pneumopathie
interstitielle sont associées à un syndrome de Raynaud, une hyperkératose
desquamante et fissurée des mains, et des anticorps anti-synthétases.
Il n'y a en principe au cours des myosites ni
atteinte rénale ni atteinte neurologique.
B
- Signes cliniques des dermatomyosites
Dans la dermatomyosite,
l’atteinte des muscles est associée à une éruption qui siège avec prédilection
aux membres et au visage. L'éruption peut exceptionnellement précéder
l'atteinte musculaire. Il s’agit le plus souvent d’un œdème des paupières
associé à un exanthème couleur lilas, étendu en lunettes au pourtour orbitaire.
L'éruption se propage souvent sur les zones découvertes de la peau, en traînées
erythémateuses ou lilacées le long des tendons extenseurs des doigts et sur la
peau des articulations des
phalanges et des coudes. Des papules de Gottron, violacées, peuvent être
observées à la face dorsale des phalanges, ainsi qu'une inflammation du
pourtour de la cuticule des ongles qui est soulevée et enflammée (signe de la
manucure).
Un syndrome de Raynaud survient dans 10 à 15% des
cas.
Une calcinose se révèle cliniquement par des nodules
durs sous-cutanés pouvant se fistuliser à la peau et laisser sourdre un liquide
crayeux. Les calcifications sont aisément visibles sur les radiographies, sous
la peau, au seion des muscles, au voisinage des articulations. La calcinose est
surtout fréquente chez l'enfant où elle survient dans 30% des dermatomyosites.
L'apparition d'une vascularite nécrosante est
exceptionnelle.
Les myosites peuvent être associées au lupus
érythémateux disséminé, à la sclérodermie et au syndrome de Gougerot-Sjögren
et, dans 15 à 20 % des cas, à un
cancer viscéral profond dont elles peuvent révéler l’existence.
Figure 1 : Signes cliniques
de la dermatomyosite.
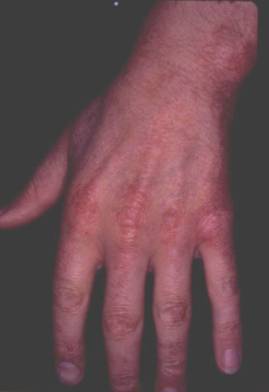
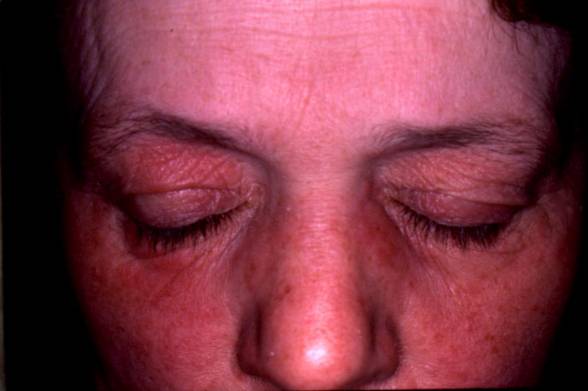
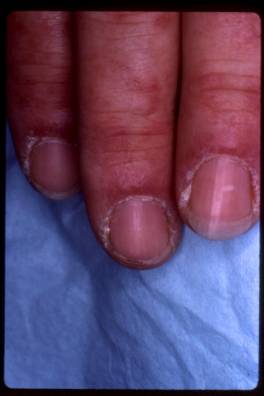
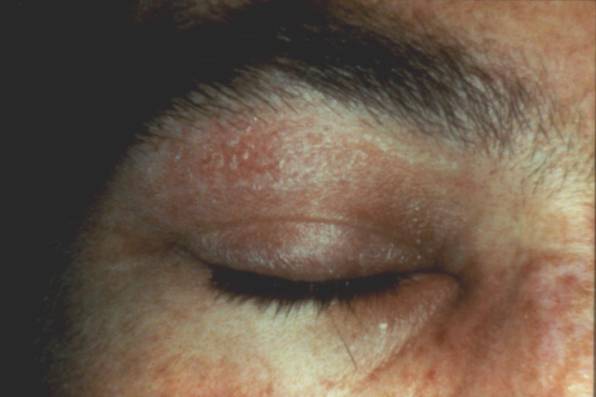
Photo : D Wallach
C
- Signes cliniques des myosites à inclusions
Les myosites à inclusions
sont des maladies chroniques entraînant un déficit et une atrophie musculaires
progressives plus souvent asymétriques que dans les autres myopathies
inflammatoires.
D - Examens complémentaires au cours des myosites
D – 1 - Les enzymes musculaires
l'
élévation des enzymes musculaires dans le plasma traduit la lyse des muscles
striés: ASAT, ALAT, LDH (lactico-déshydrogénase), aldolase, et CPK
(créatine-phosphokinase), qui est la plus spécifique. L'étude des isoformes de
la CPK est inutile. En revanche, la troponine et la MLC-1 (Cardiac myosin light
chain 1) élevées au cours de la nécrose myocardique, sont utiles au diagnostic
différentiel avec l'infarctus du myocarde.
D – 2 – La créatinurie
La lyse musculaire se traduit
aussi par une créatinurie élevée
(0,50 à 1 g/24 heures).
D – 3 – Le syndrome inflammatoire
Le syndrome inflammatoire se
traduit par une accélération de la vitesse de sédimentation, une augmentation
de la CRP plasmatique et une hyperleucocytose.
D – 4 - Les auto-anticorps
Les auto-anticorps sont peu fréquents, mais
certains peuvent apporter une aide considérable au diagnostic: Non pas tant les
facteurs rhumatoïdes que l'on observe dans 20% des cas sans qu'ils aient la
moindre spécificité pour ces maladies. Ni certains anticorps anti-nucléaires
comme les Ac anti-RNP, anti-SS/A, anti-SS/B rencontrés dans environ 20% des PM
et DM et présents au cours d'autres connectivites. En revanche, les Ac
anti-Jo1 observés dans 50% des myosites avec atteinte pulmonaire ont un
grand intérêt diagnostique car ils ont une grande spécificité pour les myosites
et orientent vers une pneumopathie. Les anticorps anti-Jo1 (anti-histidyl-ARNt
synthétase) font partie du groupe des anticorps anti-synthétases qui
caractérisent le syndrome du même nom. Les autres anticorps anti-synthétases
(anti-thréonyl-ARNt synthétase ou PL7, anti-alanyl-ARNt synthétase ou PL12,
anti-isoleucyl-ARNt synthétase ou OJ, anti-glycyl-ARNt synthétase ou EJ) sont
recherchés seulement dans les centres spécialisés. Les anticorps anti-PMS1,
spécifiques d'une ADN-réparase, sont très spécifiques des myosites, mais ne
sont détectés que dans environ 7% des cas. Les anticorps anti-SRP ( Protéines
assurant un Signal de Reconnaissance pour le transport de particules au sein du
cytoplasme) permettent de définir une forme clinique particulièrement grave et
résistante au traitement. L'anticorps anti-Mi-2, spécifique d'une enzyme
qui remodèle le nucléosome, est détecté essentiellement au cours des
dermatomyosites. Beaucoup moins performants pour le diagnostic, les anticorps
anti-Ku s'observent au cours des myosites associées à un lupus systémique, et
les anticorps anti-PM/Scl au cours des myosites associées à une sclérodermie.
D – 5 – L’étude anatomo-pathologique du muscle strié
En fait, l’examen déterminant est la biopsise
musculaire éventuellement orientée par des lésions myogènes découvertes à
l’EMG : les myosites auto-immunes se traduisent par une nécrose des fibres
musculaires entre lesquelles s’infiltrent des cellules inflammatoires
lymphoïdes et macrophagiques.
Au cours de la polymyosite, les infiltrats
inflammatoires entourent les zones nécrotiques et sont constitutés de
macrophages et de lymphocytes CD8+. Dans les lésions de
dermatomyosite, les infiltrats sont péricapillaires et constitués de
macrophages, lymphocytes B et CD4+.
Au cours des myosites à inclusions, les fibres
musculaires sont normales ou atrophiques, mais contiennent des vacuoles
intracytoplasmiques de quelques dizaines de microns remplies de granulations
éosinophiles correspondant à des structures tubulo-filamentaires d'environ 15
nm de diamètre visibles en microscopie électronique. Dans les formes
sporadiques on observe aussi un infiltrat de macrophages et de lymphocytes CD8+
à prédominance péricapillaire.
E - Traitements
Il est basé sur la
corticothérapie, d’abord à forte dose (1 mg/Kg/j de prednisone ou sous forme de
bolus de méthylprednisolone) dans les polymyosites et les dermatomyosites, puis
à posologie lentement dégressive. En cas de cortico-résistance ou de
cortico-dépendance, il faut associer un immuno-dépresseur tel que
l'améthopterine ou l'azathioprine pour parvenir à réduire les doses de
corticoïdes.
La myosite à inclusions sont
résistantes aux corticoïdes et aux immunosuppresseurs. Aucun traitement n'est
actuellement connu pour cette affection.
F - Physiopathologie
Elle est mal connue. La nature des infiltrats
inflammatoires suggère la prédominance d'une réaction auto-immunitaire au cours
de la polymyosite et d'une réaction TH2 au cours de la dermatomyosite. Les
auto-antigènes déclenchant la réponse auto-immune ne sont pas connus. Il est
cependant remarquable que les molécules enzymatiques (synthétases, Mi-2, PMS1)
reconnues par les auto-anticorps caractéristiques des myosites, sont toutes des
cibles du Granzyme B ou des caspases.
III – La connectivite mixte de Sharp
Elle est définie par la présence dans le sérum, d’Ac anti-RNP, et associe:
- un syndrome de Raynaud,
- des doigts "boudinés",
- des arthralgies,
- des myalgies,
- une dysphagie.
Le pronostic en est en principe bénin. Il s'agit cependant d'une forme clinique bénigne de LED, qui peut à tout moment de son évolution, se tranformer en LED authentique avec tout son cortège de complications possibles.
IV – La Périartérite noueuse
Décrite par KUSSMAUL et MAIER (1866), elle survient le plus souvent chez les adultes de 40 à 60 ans, répartie également entre les deux sexes. La PAN est une vascularite qui touche les artères de petit et moyen calibre selon une topographie segmentaire. Sa prévalante en Europe occidentale est d'environ 5/100 000, et son incidence de moins de 1 habitant pour 100 000 et par an.
A - Signes
cliniques
Comme dans toutes les connectivites, le diagnostic est aisé quand le
tableau clinique associe plusieurs signes, et difficile quand la maladie est
mono-symptomatique.
Les signes généraux sont
très fréquents (70% des cas) et associent de la fièvre, une profonde altération de l’état général avec
amaigrissement intense et rapide, liée à une fonte musculaire.
Les myalgies (50% des cas), d'horaire
inflammatoire, sont agravées par la palpation des masses musculaires, mais ne
correspondent pas à une véritable myosite car elles ne sont pas associées à une
élévation des enzymes musculaires.
Les arthralgies de type inflammatoire
touchent les articulations des épaules, genoux, plus rarement les petites
articulations. Elles ne se compliquent ni de déformations ni de destructions
articulaires.
Les signes neurologiques
correspondent le plus souvent à une atteinte asymétrique des nerfs
périphériques (60% des cas). Il s'agit d'une multinévrite sensitivo-motrice
douloureuse et paresthésiante, avec abolition des réflexes ostéo-tendineux dans
les territoires correspondants qui sont parfois le sièges d'oedèmes
segmentaires transitoires. L'EMG permet de confirmer le diagnostic en montrant
des signes neurogènes avec diminution des potentiels d'action moteurs et
sensitifs. Les atteintes du système nerveux central se traduisent par une
comitialité ou la survenue de déficits moteurs dont la topographie dépend de
celle de la vascularite cérébrale. Le liquide céphalo-rachidien est en principe
normal. L'IRM montre, au sein de la substance blanche, des hypersignaux en T2.
Les signes cutanés les
plus caractéristiques sont des nodules hypodermiques, souvent le long du trajet
des artères des membres. On peut observer aussi un purpura pétéchial infiltré,
et une vascularite nécrosante des extrémités, notamment au pourtour des ongles,
qui peut entraîner des ulcérations voire une gangrène. D'autres manifestations
cutanées, plus ares, sont cependant très évocatrices: le “livedo racemosa” qui
se caractérise par des marbrures ou le "livedo reticularis" qui se
traduit par un réseau de lividités.
L'atteinte rénale se
traduit par une hypertension artérielle fréquente au cours de la PAN (20 à 30%
des cas). Elle consiste en une néphropathie ischémique entraînant une
insuffisance rénale dont le pronostic peut être très sévère. L'artériographie
montre fréquemment la présence de micro-anévrysmes, et la ponction-biopsie
rénale témoigne de l'existence de lésions artériolaires caractéristiques.
Les douleurs abdominales apparaissent
dans 15% des cas. Elles sont dues à la vascularite dans le territoire de
l'artère mésentérique supérieure et peuvent simuler une urgence chirurgicale. L'artériographie mésentérique supérieure révèle des micro-anévrysmes et
des sténoses vasculaires. Cette vascularite peut entraîner des
ulcérations du tube digestif, voire des perforations viscérales.
L'atteinte cardiaque consiste
en une myocardite liée à la vascularite coronaire.
Une orchite très inflammatoire et très
douloureuse, survient exceptionnellement mais est très suggestive du
diagnostic.
NB: on distingue habituellement de
la PAN, une autre forme de vascularite, la micropolyangéite (MPA) qui
ressemble beaucoup à la PAN, mais s'en distingue par différents signes:
-
on peut observer dans la MPA une atteinte pulmonaire qui
n'existe pas dans la PAN: dyspnée, toux, hémoptysies avec infiltrats
mulmonaires à la radiographie et à la tomodensitométrie.
-
L'atteinte rénale de la MPA n'est pas artériolaire mais
glomérulaire.
-
Les angiographies ne montrent pas de micro-anévrysmes dans
la MPA.
Figure 1 : périartérite
noueuse avec purpura palpable sur les cuisses.
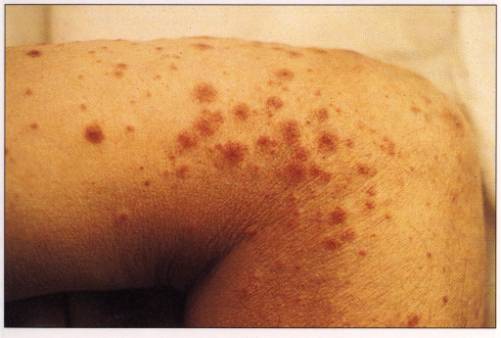
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
Figure 2 : nodules
sous épidermique au cours d’une périartérite noueuse, vascularite avec rupture
de la limitante élastique interne (Thr : thrombus et FN : nécrose
fibrinoïde), infiltrat à polynucléaires neutrophiles (PNN) et éosinophiles.
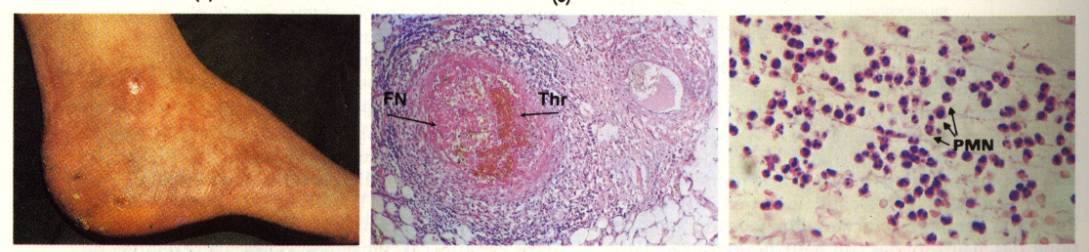
Photo :
Immunologie, 1990, I. Roitt, Ed Pradel
B - Signes
biologiques
- La PAN
est la seule connectivite qui ne s’accompagne pas d’anticorps antinucléaires ni, en principe, d'anticorps
anti-cytoplasme de polynucléaires (ANCA). Dans 50% des cas de MPA on
observe des ANCA spécifiques de la myéloperoxydase.
-
L’examen qui permet de faire le diagnostic de PAN est la biopsie d’un nodule. On peut alors visualiser une artérite
touchant un vaisseau de moyen calibre, comportant un épaississement de
l’intima, une nécrose éosinophile de la média avec une rupture de la limitante
élastique interne, et un infiltrat péri-adventitiel comportant des macrophages,
des lymphocytes et des polynucléaires neutrophiles et éosinophiles.
Lorsqu'
aucun nodule n'est palpable, une biopsie neuro-musculaire doit être réalisée,
guidée par un EMG des membres ou, si l'EMG est normal, dans le territoire du
sciatique poplité externe. Un aspect de vascularite peut alors être observé
autour d'une artère musculaire ou autour d'un rameau nerveux.
- La NFS
montre une hyperleucocytose au-dessus de 10 000 leucocytes /mm3 avec une hyperéosinophilie > 1000 /mm3.
- La VS et la CRP sont élevées
- En cas d’atteinte rénale, la créatinine sérique est élevée,
une protéinurie et une hématurie microscopique apparaissent.
-
Il est fréquent que l’on retrouve chez les patients, la
présence d’un antigène HbS
circulant, ou du moins les anticorps
anti-HbS correspondants.
C -
Physiopathologie
Il est
vraisemblable que la PAN résulte d'une stimulation antigénique chronique, par
exemple par un agent infectieux, et que des complexes antigène-anticorps
déposés au sein de l'endothélium déclenchent le processus inflammatoire qui
caractérise la vascularite.
D - Evolution
et traitement
En l’absence de traitement, la maladie peut évoluer
vers la défaillance viscérale et la mort. Il existe de rares cas de guérison
spontanée, et de nombreux cas de longue survie grâce à la corticothérapie qui
reste le traitement de base, éventuellement sous forme de bolus dans les formes
aiguës. Une immuno-suppression (alkylants, azathioprine, améthoptérine) doit
être associée en cas d'efficacité insuffisante de la corticothérapie. Les
formes associant une infection par le virus de l'hépatite B doivent être
traitées en outre par des anti-viraux et échanges plasmatiques. Le traitement
sera le plus souvent maintenu à vie, à des doses progressivement décroissantes
jusqu'au seuil de réapparition du syndrome inflammatoire ou des symptômes.
V – La Maladie de Wegener
La maladie de Wegener est une granulomatose bi-polaire touchant
l'appareil respiratoire et les reins, dont souffrent environ deux personnes
pour 100 000 habitants en occident. Moins de 0,3 cas pour 100 000 habitants
apparaissent chaque année. Cette affection est aussi fréquente chez l'homme que
chez la femme, survient à tout âge, avec une prédominance vers 40-50 ans, et
environ 10% de formes pédiatriques.
A - Signes
cliniques
-
Atteinte
respiratoire: alors qu'en général, au cours
des connectivites, c'est le poumon qui est préférentiellement touché, la
vascularite de Wegener commence souvent par une atteinte des voies aériennes
supérieures: sinus et cavités nasale (70% des cas).
o Sinusite et rhinite: Le malade se plaint d'obstruction nasale chronique,
de rhinorrhée séreuse, parfois sanglante, parfois surinfectée et purulente. La
sinusite se traduit par des douleurs intenses de la face et du crâne. Ces
signes ne régressent pas, même sous l'effet d'une antibiothérapie adaptée aux
germes. L'ORL consulté observe une muqueuse nasale friable et hémorragique, et
parfois une otite moyenne. Le caractère nécrosant est typique de la vascularite
de Wegener et se concrétise par des perforations de la cloison nasale et des
destructions osseuses sinusiennes visibles à la tomo-densitométrie.
o Atteinte pulmonaire: elle se traduit par une toux, une expectoration
souvent hémoptoïque, des douleurs thoraciques et une dyspnée.La
tomo-densitométrie montre des opacités nodulaires et des infiltrats parfois excavés.
La fibroscopie bronchique ne montre qu'une inflammation bronchique non
spécifique. Le lavage broncho-alvéolaire rapporte surtout des polynucléaires
neutrophiles, témoignat aussi d'une inflammation non spécifique. Lorsque la vascularite de Wegener se
présente initialement sous la forme d'une maladie pulmonaire, le diagnostic
différentiel entre une vascularite et une infection grave peut donc être très
difficile.
-
L'atteinte
rénale, tout en ajoutant à la gravité
de la maladie, facilite son diagnostic lorsqu'elle est associée aux atteintes
respiratoires. Présente dans plus de la moitié des cas, elle correspond à une
glomérulonéphrite nécrosante. Elle se traduit par une hypertension artérielle,
une protéinurie, une hématurie et une élévation de la créatinine sérique. Elle
peut évoluer très vite vers l'insuffisance rénale aiguë avec anurie et
nécessite un traitement d'urgence. L'examen histologique montre une
glomérulonéphrite nécrosante segmentaire et focale avec prolifération
extra(capillaire segmentaire et focale dont l'extension définit la gravité de
l'atteinte.
-
D'
autres manifestations cliniques,
moins caractéristiques de la maladie de Wegener, sont cependant fréquemment
rencontrées:
o Les atteintes cutanées sous forme de purpura infiltré et de nodules hypodermiques,
peucvent donner le change avec une PAN
o Les arthralgies et des myalgies ressemblent à celles que ressentent les patients au
cours de toutes les vascularites.
o L'atteinte oculaire XXX
o
Une
multinévrite peut aussi compliquer la
vascularite
Figure 1 : Critères
diagnostiques de la maladie de Wegener.

B - Signes
biologiques
-
Les critères histologiques de vascularite nécrosante sont
caractéristiques mais parfois difficiles à obtenir ou à interpréter.
-
Le meilleur signe biologique est la présence
d'anticorps anti-cytoplasme de poynucléaires (ANCA) dans le sérum. Ces
anticorps sont détectés par immuno-fluorescence indirecte sur frottis de
polynucléaires neutrophiles où ils déterminent une fluorescence cytoplasmique.
Ils sont présents dans plus de 90% des cas de la maladie. Il faut ensuite
déterminer leur spécificité par ELISA. Il existe des ANCA anti-protéinase 3,
anti-myéloperoxydase, anti-cathepsine, anti-lactoferrine. Les ANCA
anti-protéinase 3 (PR-3) ont une
spécificité de 100% pour la maladie de Wegener. Les autres ANCA peuvent être
détectés dans de nombreuses connectivites et autres maladies inflammatoires
comme les maladies inflammatoires chroniques auto-immunes de l'intestin
–maladie de Crohn, rectocolite hémorragique).
Figure 2 : Anticorps
anti-cytoplasme des polynucléaires.
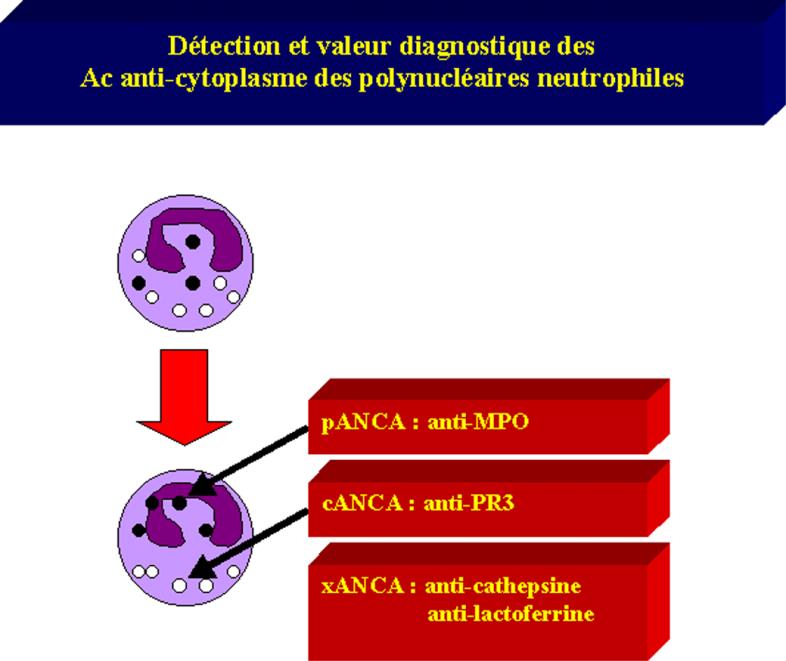
Maladie |
Aspect Fluo |
Antigène |
Fréquence |
Wegener |
C |
PR3 |
85% |
|
PAN |
C,P |
MPO,PR3 |
Rare |
|
Artérite à
cellule géante |
P |
MPO |
Rare |
|
Vascularites
médicamenteuses |
P |
MPO |
Rare |
|
Polyarthrite Rhumatoïde |
P,X |
MPO, Lactoferrine, cathepsine |
Rare |
|
LED |
X |
Lactoferrine, cathepsine |
Rare |
|
Maladie de
Crohn |
P,X |
MPO, Lactoferrine, cathepsine |
10-40% |
|
RCH |
P,X |
MPO, Lactoferrine, cathepsine |
40-80% |
|
Cholangite
sclérosante |
P,X |
MPO, Lactoferrine, cathepsine |
65-85% |
Figure 3 : Aspect des
anticorps anti-cytoplasme des polynucléaires.
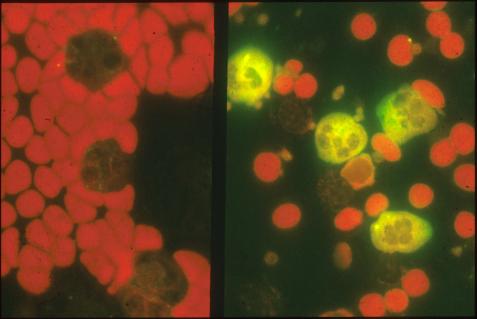
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
C -
Physiopathologie
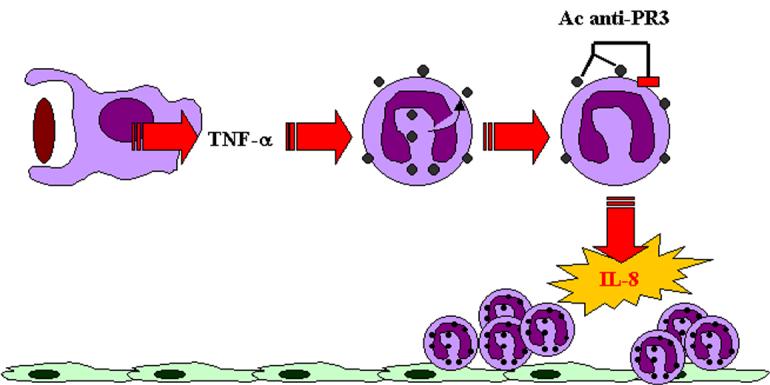
Au décours d’une infection des
voies respiratoires, l’agent infectieux stimule les macrophages et entraîne la
production de TNF-a. Cette cytokine pro-inflammatoire agit sur les
polynucléaires neutrophiles et favorise l’expression membranaire des granules
cytoplasmiques du polynucléaires. Les antigènes reconnus par les ANCA se
retrouvent ainsi exposés à la surface des polynucléaires. Les ANCA se fixent
par leurs fragments Fab sur la protéinase-3 et par leur fragment Fc sur le RfcgRI.
Cette double fixation entraîne l’activation et la libération d’IL-8 par le
polynucléaire. L’IL-8 est une cytokine douée d’un pouvoir chimiotactique
puissant sur les polynucléaires neutrophiles. La libération massive d’IL-8 entraîne
donc un afflux massif de polynucléaires qui seront alors responsables de
l’atteinte vasculaire.
Figure 4 : Physiopathologie
de la vascularite de Wegener.
B -
Traitement
En l'absence de traitement, la maladie
est presque constamment mortelle. En cas d'atteinte rénale, si le traitement
est différé, une insuffisance rénale aiguë définitive peut survenir: autant
dire que le traitement de la maladie de Wegener est un traitement puissant et
immédiat, associant d'emblée une corticothérapie et un immunosuppresseur
(alkylant), éventuellement sous forme de bolus. Le traitement sera le plus
souvent maintenu à vie, à des doses progressivement décroissantes jusqu'au
seuil de réapparition du syndrome inflammatoire ou des symptômes.
V – Les Sclérodermies
A - Signes
cliniques
Le terme de sclérodermie désigne des affections caractérisées par un épaississement fibreux de la peau et représentent deux grandes variétés d’affections :
- Les sclérodermies cutanées pures dans lesquelles les lésions sont limités à la peau
- Les sclérodermies généralisées ou systémiques dans lesquelles la fibrose touche également les vaisseaux et peut atteindre différents organes : tube digestif, poumon, rein et cœur.
Les sclérodermies sont des maladies rares, deux fois plus fréquentes chez la femme que chez l’homme et qui surviennent en général vers 40 ans.
- Les sclérodermies cutanées entraînent une sclérodactylie et peuvent s’étendre aux membres et au visage où elles limitent le degré d’ouverture de la bouche. Elles sont toujours associées à un syndrome de Raynaud qui précède parfois de 10 à 15 ans les manifestations cutanées. Ces formes, qui représentent environ la moitié des sclérodermies systémiques sont d’évolution lente et relativement bénigne. Dans ce groupe, on a individualisé le syndrome CREST qui associe de façon plus ou moins complète : des calcifications sous-cutanées, syndrome de Raynaud, atteinte oesophagienne (visible au radio-cinéma), sclérodactylie et télangiectasies.
- Les sclérodermies systémiques sont les plus graves car elles n’atteignent pas seulement la peau, mais aussi certains viscères comme le rein, les poumons, le cœur et le cerveau.
Les
syndromes CREST ne mettent pas en jeu le pronostic vital mais peuvent entraîner
une impotence fonctionnelle très grave. Au contraire, les sclérodermies
systémiques sont souvent peu gênantes sur le plan fonctionnel, mais elles
peuvent évoluer vers la mort.
Figure 1 : Sclérodermies
cutanées.
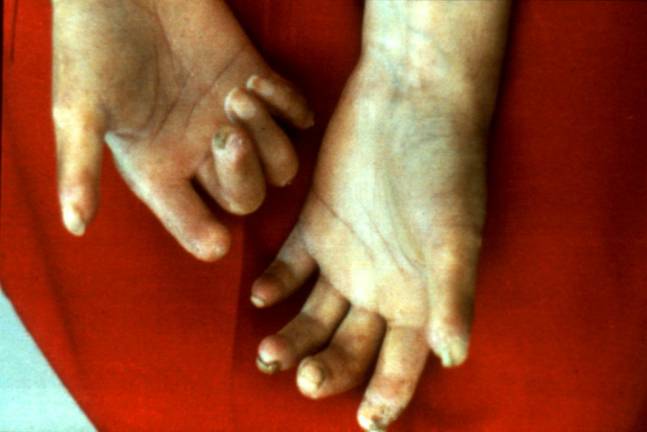
Photo : B Weill
Figure 2 : Anomalies capilaires
au cours des sclérodermies.
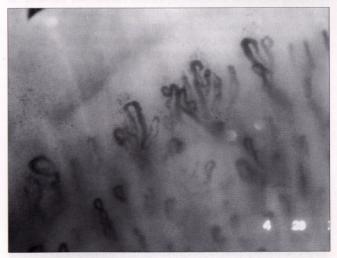
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
Figure 3 : Atteinte
oesophagienne des sclérodermies.
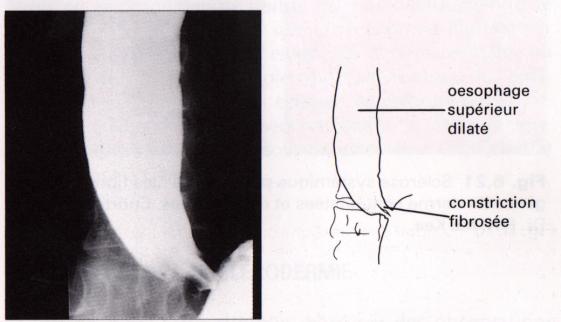
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
Figure 4 : Calcification
sous cutanées des sclérodermies.
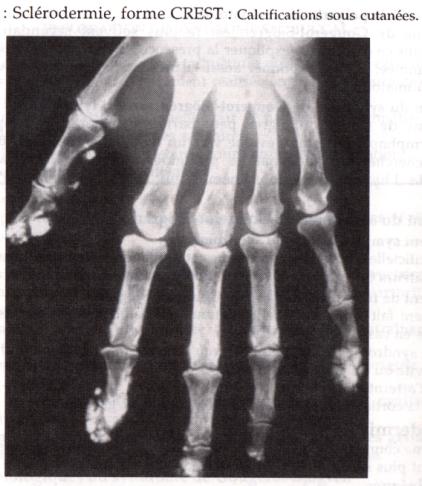
Photo :
Rhumatologie, 1992, S. Perrot, Ed Med-Line
B - Signes biologiques
En dehors des signes propres aux atteintes viscérales, les sclérodermies sont caractérisées par la présence de certains auto-anticorps. Des auto-anticorps antinucléaires sont détectés dans 60 % des cas, et des facteurs rhumatoïdes dans 30 % des cas. Certains anticorps antinucléaires permettent de définir la forme clinique de la maladie :
- on trouve en effet des Ac anti-centromères dans 85 % des syndromes CREST
- des anticorps anti-Scl70 dans 50 % des sclérodermies systémiques.
(cf chapitre des auto-anticorps non spécifiques d’organes).
C - Evolution
et traitement
L’évolution dépend de l’extension viscérale. Il n’ y a pas de traitement vraiment efficace. Les corticoïdes sont généralement contre-indiqués, surtout en cas d’atteinte rénale.
VI – Le syndrome de Gougerot-Sjögren
A -
Manifestations cliniques
Le syndrome de Gougerot-Sjögren (SGS) ou syndrome sec se caractérise par un tarissement des sécrétions salivaires, lacrymales, nasales et vaginales. Dans plus de la moitié des cas, il est associé à un rhumatisme inflammatoire. L'association la plus fréquente est celle d'une polyarthrite rhumatoïde (25 à 50% des cas), puis d'une sclérodermie (5 à 15% des cas), du lupus systémique (5 à 15% des cas), de la polymyosite 6% des cas) ou d'une polyarthrite de nature indéterminée (2% des cas). La maladie atteint souvent les femmes d'âge moyen. Les signes articulaires peuvent précéder la xérostomie et la xérophtalmie.
La sécheresse oculaire, soupçonnée après l'interrogatoire du malade est confirmée par le test de Schirmer, le test au Rose Bengale et surtout par l'examen de la cornée au bio-microscope qui montre une kératite filamenteuse.
L'atteinte
des glandes salivaires n'apparaît pas toujours en même temps que l'atteinte
oculaire. Elle semble moins fréquente. On observe dans 50% des cas un
gonflement des parotides, généralement bilatéral et symétrique, plus souvent
transitoire que chronique, précédant la xérostomie. La dysphagie et les
gastralgies sont fréquentes.
Figure 5 : Infiltration des
glandes salivaires au cours du syndrome de Gougerot-Sjögren.
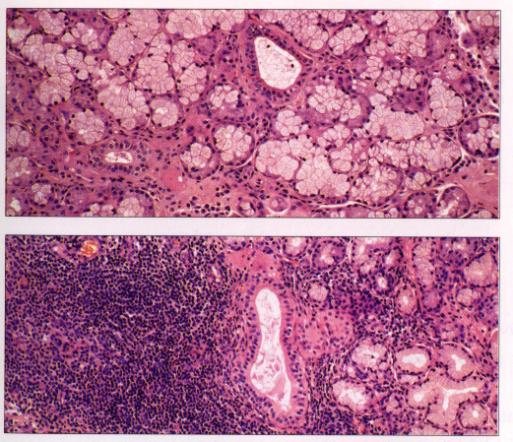
Photo : Immunologie clinique, 1991, J. Brostoff, Ed DeBoeck Université
Les connnectivites éventuellement associées au syndrome de GS peuvent être accompagnées des atteintes viscérales qui ont été étudiées dans les chapitres correspondants. Le syndrome de GS peut être accompagné d'autres maladies auto-immunes telles qu'une hépatite chronique, une cirrhose biliaire primitive, une thyroïdite, une anémie hémolytique à auto-anticorps, une vascularite, une myasthénie.
On distingue le syndrome sec primaire isolé, et le syndrome sec secondaire associé à une connectivite ou à une autre maladie auto-immune dont les manifestations cliniques sont parfois au premier plan. En fait, le syndrome est rarement isolé, puisqu'il est souvent associé à un syndrome de Raynaud, à un purpura d'origine vasculaire, à une polyadénopathie, à une splénomégalie, à des signes de myosite ou enfin à une atteinte spécifique rénale ou pulmonaire.
Plus de 60% des malades souffrent d'accidents allergiques médicamenteux.
Le SGS primaire peut se compliquer de localisations tumorales "pseudolymphomateuses" ou de véritales lymphomes malins tels qu'une macroglobulinémie de Waldenström.
B -
Diagnostic biologique
- Anticorps antinucléaires (AAN) : l'immunofluorescence sur coupes de foie de rat permet de détecter des AAN chez la moitié des patients atteints de SGS. En l'absence de LE, on ne trouve pas d'Ac anti-ADNn. Les Ac anti-ECT sont rencontrés avec la fréquence qui caractérise la connectivite associée au SGS. Des Ac anti-SS-B (ou anti-Ha ou anti-La) sont détectés dans 80% des SGS primaires; 70% des patients ont aussi des Ac anti-SS-A (ou anti-Ro). Les Ac anti-SS-B ne sont habituellement pas observés dans le SGS secondaire en dehors de l'association avec un LED. En revanche, les Ac anti-SS-A sont observés dans 30% des SGS secondaires et dans 20% des LED sans SGS. On détecte couramment des Ac anti-SS-A en l'absence d'Ac anti-SS-B, mais des Ac anti-SS-B sont exceptionnellement présents sans Ac anti-SS-A.
- Autres auto-anticorps : Des facteurs rhumatoïdes sont détectés par le test au latex (Singer-Plotz) ou le test ELISA dans 60% des cas. En cas d'association à une polyarthrite rhumatoïde, le test de Waaler-Rose est en général positif. La fréquence de positivité d'au moins l'un des trois tests atteint alors 90%.
De nombreux autres auto-Ac peuvent être observés au cours de cette maladie : des Ac anti-thyroïde (anti-thyroglobuline et anti-thyroperoxydase), Ac anti-cellules pariétales de l'estomac, Ac anti-muscle lisse, et anti-mitochondries de type 2. Des Ac anti-hématies peuvent être responsables de la positivité du test de Coombs, mais les anémies hémolytiques auto-immunes sont rares.
- Modifications des immunoglobulines :
- Immunoglobulines salivaires : la salive ne contient normalement comme immunoglobulines que des IgA sécrétoires. En cas de SGS cependant, des IgM puis des IgG peuvent apparaître; les IgM sont les plus caractéristiques de la maladie. Bien que le dosage lui-même soit aisé par immunodiffusion radiale ou par néphélométrie, le prélèvement salivaire requiert un cathétérisme du canal de Sténon qui est très douloureux. Le simple recueil de la salive intra-buccale donne des résultats aléatoires car la moindre affection bucco-dentaire se traduit par l'apparition d'IgG localement.
- Immunoglobulines sériques : l'hypergammaglobulinémie est constante. La diminution des immunoglobulines sériques fait redouter la survenue d'un lymphome et doit faire rechercher l'apparition d'une immunoglobuline monoclonale par immuno-fixation ou immuno-électrophorèse.
C -
Diagnostic anatomo-pathologique
Seul l'examen anatomo-pathologique d'une glande exocrine permet de poser avec certitude le diagnostic de SGS. Les biopsies peuvent porter sur la muqueuse nasale (où elles sont douloureuses et parfois hémorragiques) ou de préférence sur la muqueuse labiale où se trouvent de nombreuses glandes salivaires accessoires. Il ne faut pas biopsier les glandes salivaires principales qui se fistulisent facilement. En cas de SGS on note une régression canalaire des acini, une fibrose péri-acineuse et une infiltration lymphoïde des glandes.
D -
Traitement
- Traitement local :
- Larmes artificielles, ou gel-larmes, ou inserts pour la xérophtalmie.
- Salive artificielle (souvent mal supportée) pour la xérostomie.
- Traitement général :
C'est celui de la connectivite associée au SGS secondaire, en tenant compte de la fréquence des allergies médicamenteuses. L'hydroxychloroquine (Plaquénil®) est souvent efficace, associée à de petites doses de prednisone (5 à 8 mg par jour).